The complexity of chemistry stems from our inability to universally determine all the relevant dynamical processes involved in chemical transformations with an accurate accounting of electronic structure. Even relatively “simple” chemical reactions can be extremely complex when diverse reaction pathways connect many transient species. Robust methods are needed to predict how a set of reactants undergoes sequential, branching reactions, passing through many transition states and transient species, to reach a final set of stable products. The dynamics of these species undergoing chemical changes are governed by the complex topology of the force field. The usual approach for explaining or predicting overall chemistry is to assume a mechanism of elementary reactions and energy transfer processes, and thus involves a large number of differential equations with ad hoc parameters. The main problem of this approach is determining all the relevant processes and parameters. However, even if one could know all the elementary processes to include, accurately determine all the parameters, and perform the numerical integrations of the huge number of coupled differential equations for the duration of the overall reaction, understanding how the chemistry evolves in such models is far from simple, and essentially impossible for practical applications. New approaches are needed.
Complex chemistry is the current salient challenge in chemical kinetics. We need robust methods for determining the critical behaviors of complex overall reactions if we are to develop predictive theories. We approach this problem with the idea that it is not necessary to describe or even know all the details, only those that are directly involved in the pathway from reactants to products; that is, the dominant route through the complex topology of peaks and valleys of the force field for a given set of experimental conditions. We also consider it essential to understand the role of fluctuations in the reaction rate that can be induced by the energetic environment and the many intermediates in combustion processes. If this can be accomplished, it may be possible to design external protocols to control the products of chemical reactions and optimize these protocols to be energy efficient.
Our central objective is to describe, predict, and control complex chemical behaviors. This is a challenging undertaking, and will require an innovative new framework. Thus, a synergistic approach is proposed that integrates the full range of rigorous fundamental theoretical methods to obtain effective predictive methods for complex chemistry.
Team Members: Ryan Mohr, Emir Yasun
Funding: Army Research Office grant ARO-MURI W911NF-14-1-0359
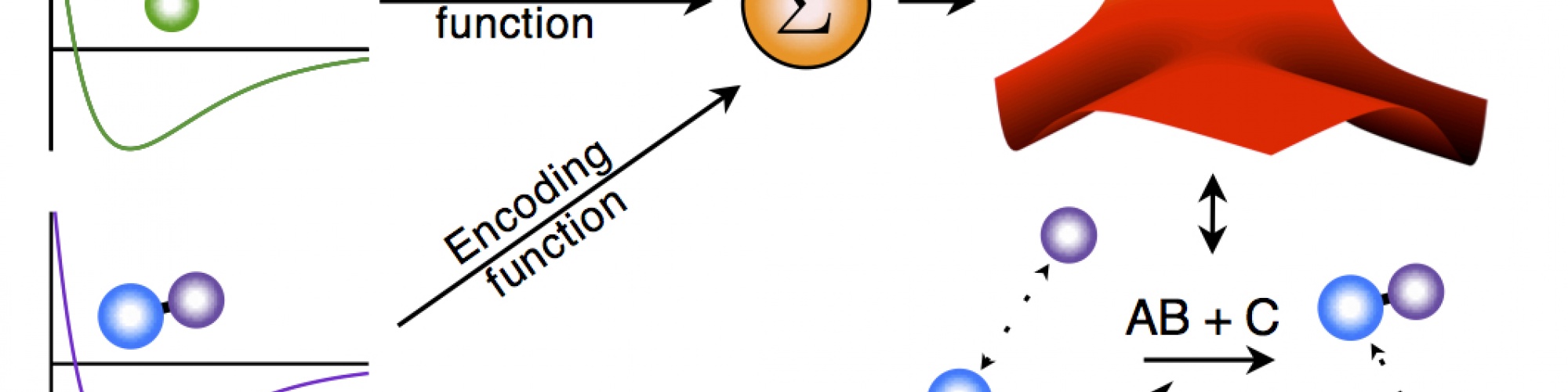
Programmable Potentials
Chemical reactions are governed by the electronic structure of the system. However, getting a full potential, for even a moderately sized system, using ab initio methods is intractable; naive methods scale exponentially with the number of electrons. The best we can do is to find an approximate potential that respects the physics of the problem.
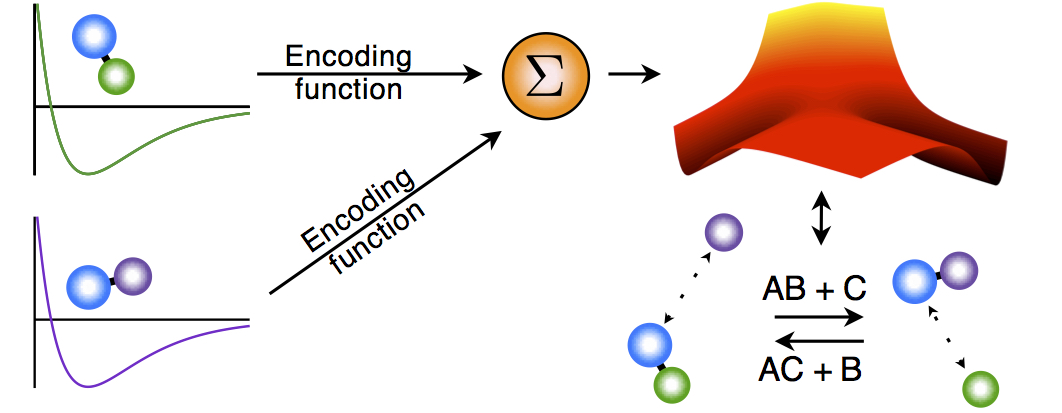
We are currently building a modeling methodology that takes as an input experimental observations and ab initio calculations for pairwise molecules and combines them to form a global potential that governs the reaction. The ab initio calculations for the pairwise interactions can be found with standard methods. The idea is to combine these pairwise potentials into a global potential. We do this by using the observed physics of the system to generate logic rules which are then translated into smooth encoding functions that modify each pairwise potential. The effect of the encoding function is merely to turn its associated pairwise potential "on" and "off" depending on the molecular configuration of the system. The resulting global potential has the form of a linear combination of the pairwise potentials, except that the "coefficient" of each pairwise potential is one of the encoding functions.

Programmable Potentials methodology
|
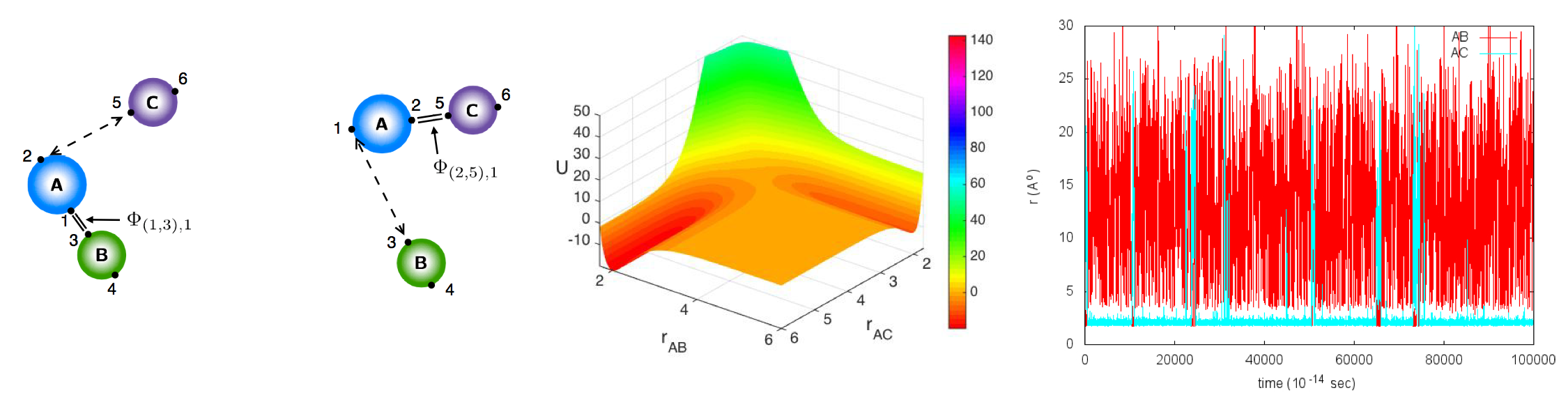
Biased chemical reaction, potential energy surface, and LAMMPS simulation
|

Simple model of DNA transcription
|
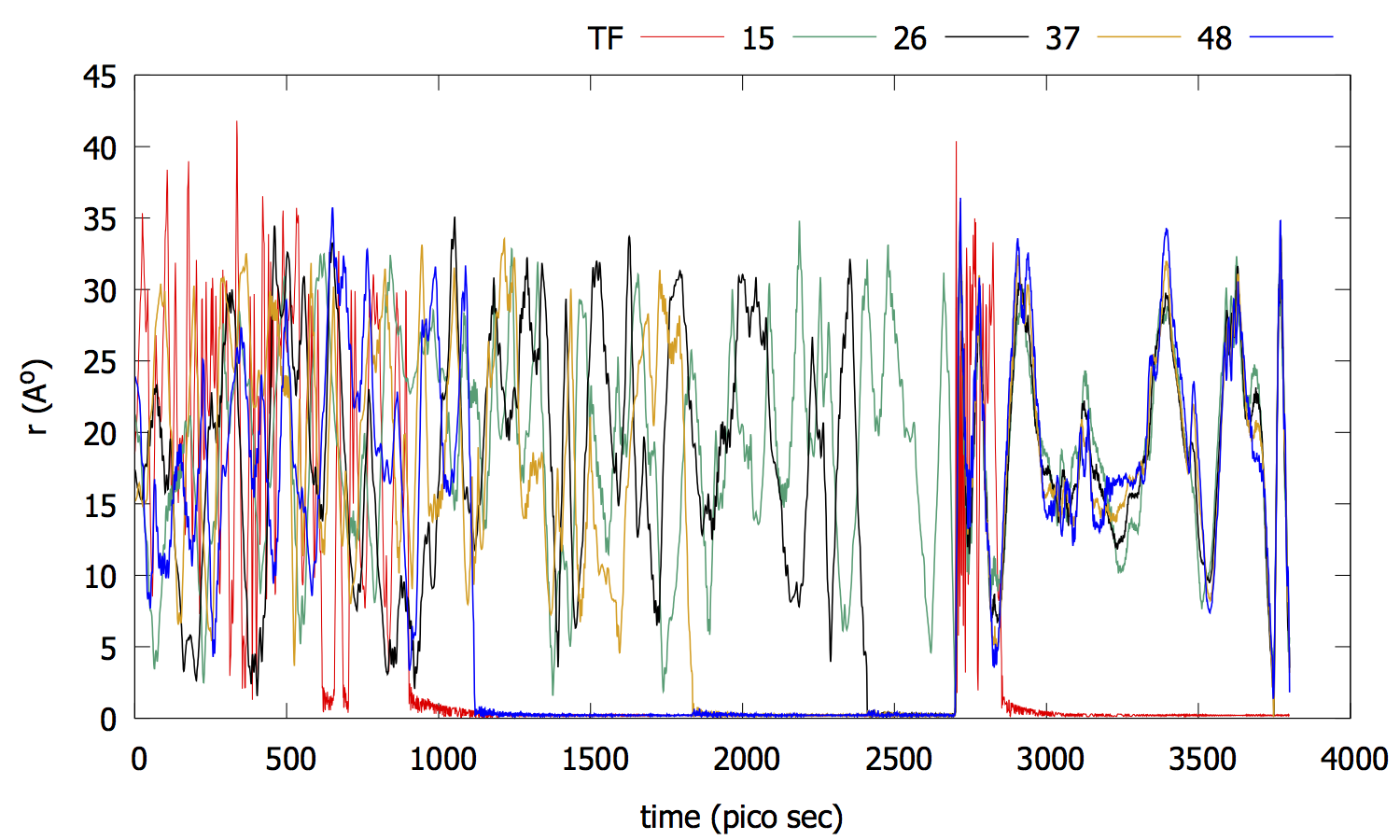
LAMMPS simulation for DNA transcription potential
|
Experimental Verification
We are currently in the process of experimentally verifying the Programmable Potentials methodology. Emir is running DNA hybridization experiments which will then be compared to molecular dynamics simulations driven by the analytic potential developed by Ryan using the Programmable Potentials methodology.